Rhona O’Donnell Senior Director, Data Management & Clinical Risk Management – ICON
Lalit Pai Senior Vice President & Head of Global Biometrics – ICON
It is generally agreed that the pandemic accelerated the adoption of decentralized and hybrid clinical trials and according to many commentators they are here to stay. Many countries in Asia appear to be open to the use of these models, however regulatory authorities are looking to other regions to see how they are responding before issuing new guidance. Pharma and biotech organizations in Asia are looking to CROs for practical guidance on how to conduct these clinical trials and exploring what is possible in different countries. Anecdotally, it appears that investigative sites are already getting ready for the changes they will need to make to manage clinical trials that have decentralized components, such as some patient visits being conducted at home or in a convenient location.
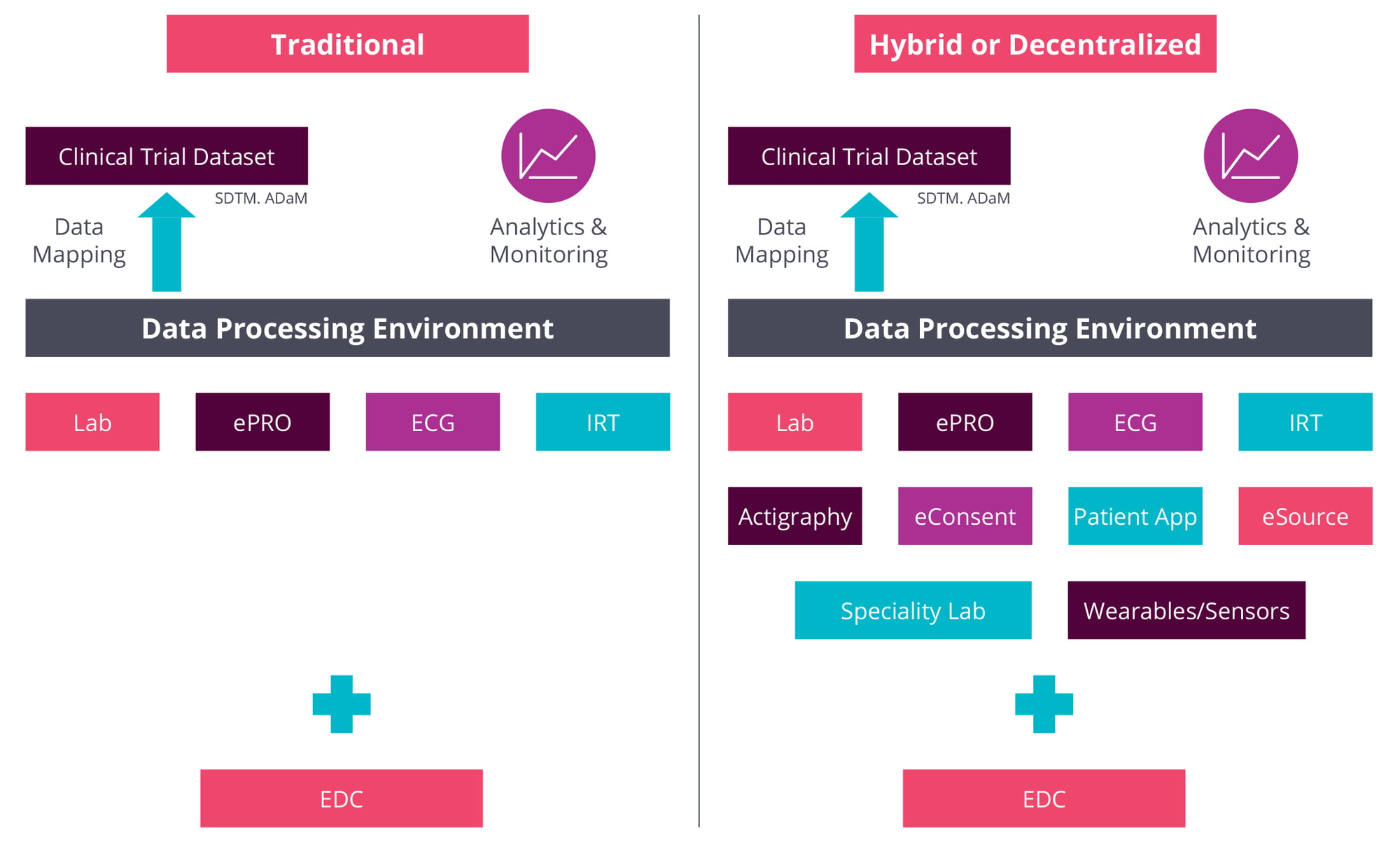
Regardless of where the clinical trial is conducted, as the shift towards decentralized and hybrid clinical trials expands the number and variety of available data sources, the volume, diversity and complexity of accumulated data have increased in parallel. Source diversity enables treatment effects to be assessed from different perspectives and in different settings, with more emphasis on real-world quality-of-life measurements. Ultimately, it nudges the trial closer to how a drug might perform in a broader patient population.
These benefits align with growing demand for demonstrable value in healthcare systems, however they do not come without challenges. The sheer volume of data coming out of clinical trials is not new but the range and heterogeneity of data sources, call for a whole new level of oversight. As Rhona O’Donnell, ICON’s senior director, Data Management & Clinical Risk Management, explains these used to be limited, typically, to electronic data capture and anything from one to four other sources, such as laboratory and electrocardiogram data, interactive response technology and electronic patient-reported outcomes.
Decentralized trials add to that list data from specialty labs, from wearables and sensors, patient apps, actigraphy devices, eConsent processes and eSource systems.
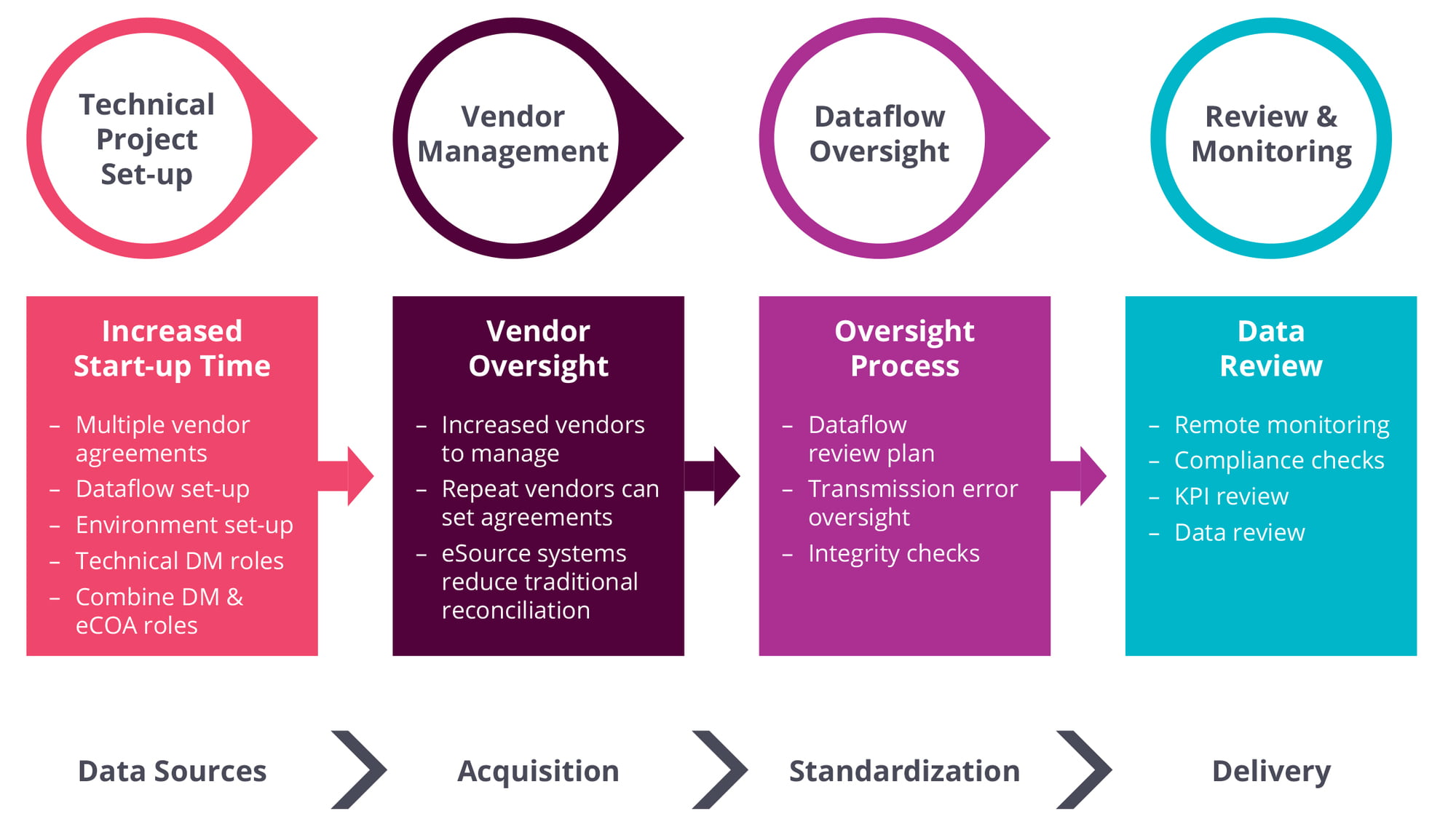
As O’Donnell notes, each of these requires a distinct process to set up the source, deliver the data, perform integrity checks, and review the data against the sources. “And you still have to have all of this ready for when the first patient is in,” she adds. “There’s much more of a technical set-up, and much more planning is required upfront.”
With established suppliers such as lab vendors, the process is facilitated by highly standardized data-transfer agreements, where “we know what data to expect”, O’Donnell continues. With new technologies such as wearables and sensors, however, vendors may have less experience of clinical trials, requiring closer attention to issues such as data reliability and ease of integration.
Moreover, observes Lalit Pai, senior vice president and head of Global Biometrics at ICON, the potential variability of data from a wider range of sources means the whole “data backbone needs to be more robust now”. Those data may include patient-generated content from different types of devices, raising data-transfer, -anonymity and -quality questions while pushing up complexity levels.
These issues must be worked out with data vendors upfront at the protocol-planning stage, O’Donnell stresses. For example, some vendors may collect “millions and millions” of patient records but provide only summarized datasets. With other vendors, “we are going to get all the records, and then we need to work out algorithms and how we’re going to derive the information.”
Clinical Trial Dataflow: Traditional vs Hybrid or Decentralized Trials
The same principles apply to ensuring the consistency and reliability of clinical-trial data generated virtually, particularly when this is done on a bring-your-own-device basis. Again, it is about defining in advance the data to be collected, organizing data collection collaboratively with the provider, agreeing associated data formats, and conducting user-acceptance testing on data-generating devices.
Pai also emphasizes the importance of contextualizing data outputs. Each study is different, whether by therapeutic area or the way the data need to be interpreted. Using their subject-matter expertise and the company’s ICONIK clinical informatics platform, ICON’s data managers can identify patterns in data from multiple sources and “look at things that don’t appear right” Pai explains.
Another important step during study set-up is defining the parameters for data review. This means drawing up a coordinated plan that establishes the various roles of medical monitors (reviewing data from a medical perspective), data managers (reviewing consistency/cleanliness; looking for patterns in the data) or clinical-data analysts (looking for outliers and trending; providing feedback to sites on data quality).
Standardization across portfolios and the industry would be a great help in this respect, Pai adds. In some areas, customers may be looking for more data variability to provide richer insights. However, breaking the mold in every data domain “is not really necessary”, Pai insists.
Better to have the right balance of standardized and non-standardized data, generating real value while facilitating activities such as downstream processing.
There may also be challenges around connectivity and device malfunction in trials relying on mobile technology “I don’t think any eCOA (electronic clinical-outcome assessment) device is going to function 100% perfectly,” O’Donnell comments. Moreover, when eCOA is used for patient questionnaires or eDiaries, these are contemp-oraneous data. If they are not collected at the time, “you can’t look back and decide how you felt two days ago”.
As Pai notes, much of this comes down to data management at device level. Each app will have an administrative layer or ‘overhead’, consisting of, for example, a function that logs every keystroke or data transfer. App/device companies need to consider the amount of overhead they incorporate (increasing required system resources and slowing down performance) in relation to the volume of data they want to capture.
“Without a certain minimum level of data logging, we will not allow the eCOA or app vendor to go ahead with the trial,” Pai comments. If an issue such as device malfunction results in data loss, then without an acceptable degree of data logging, data managers risk being left high and dry.
Data privacy, security and anonymity are also issues that require early risk assessment to identify and pre-empt any potential vulnerabilities and country specific requirements. As O’Donnell explains, there are already strong protections in place with more traditional EDC, IRT or test-laboratory systems.
Data are anonymized with no central decoding key, and only the trial sites know the patient identifier. With newer vendors, though, such as BYOD of eCOA providers, more effort may be needed to ensure the necessary firewalls are in place and anonymity is watertight.
Data integration is one more challenge amplified by the proliferation of new data sources. As soon as data are collected, they must be integrated and surfaced through the ICONIK platform, so that useable data are available both to sponsors and all the relevant teams within ICON. Here again, newer sources such as sensors or wearables may be more problematic, due to volume or variability in data formats and the data themselves.
Technology and analytics capabilities have evolved along with the expansion of data sources in clinical trials. It is not just about surfacing and aggregating the data but generating more sophisticated visualizations as a basis for meaningful extraction and observation. At the back end of the operation, Pai adds, more advanced project-management and data-integration skills are needed to handle inputs from new data vendors. Consumer-device companies, for example, may not understand so well the necessity for data validation or retention.
For service providers addressing the challenges of multiple data sources in decentralized trials, the immediate pay-off is accelerated data flow directly into data-integration systems, rather than relying on trial sites to input data in their own time. With newer sources such as eCOA, wearables or sensors giving real-time access to data, “we can review these more quickly and figure out much faster what’s going on with the trial across the board,” O’Donnell points out.
Engaging with issues raised by data-source expansion at the earliest opportunity ensures that decentralized trials not only significantly enhance the patient experience but deliver more timely, comprehensive insights to optimize study outcomes and, ultimately, product value. In a pharmaceutical market where data increasingly are the product, getting data management right from the start is a crucial part of the value equation.
Adapting Roles and Processes for Decentralized Trials: Data Management and Clinical Operations
Varying Implementation Of India'sTrial Waiver Clause
BY ANJU GHANGURDE
The new chief of the Indian Society for Clinical Research, Sanish Davis, tells Scrip how the Indian trials segment coped amid pandemic upheaval, emphasizing the need for stakeholder “sensitization and socialization” of the concepts of virtual/hybrid trials as part of efforts to mainstream these. The executive also shared his views on India’s trial waiver clause.
The Indian clinical trials sector, like many others, has had to navigate a turbulent 2020 as the pandemic forced a shift in traditional approaches to working amid lock-downs, challenges in patient mobility and re-purposing of clinical sites as COVID-19 care centers.
In a wide-ranging interview with Scrip, Dr Sanish Davis, incoming president of the Indian Society for Clinical Research (ISCR) and R&D director, Global Clinical Operations, India at Johnson & Johnson, outlined how industry kept things going in India with new interventions and remote monitoring and also tackled the complexities of audio-visual recording of the informed consent process amid pandemic-related complexities.
Dr Sanish Davis
ISCR counts among its members several large multinational firms and clinical research organizations and Davis will be at its helm through 2021-23.
Notably, while under 2% of global trials are estimated to be conducted in India, leaving a lot to be desired, the country’s trial trajectory until mid-November 2020, though subdued amid COVID-19, did not fall off the cliff and was in line with broader global trends. An analysis covering clinical trials initiated by biopharma with a confirmed start date of between January 2019 and December 2020, indicated that 2020’s total of 5,276 trials declined only 8% versus the 5,706 trials reported by Informa’s Trialtrove in 2019, despite the pandemic upheaval. (Also see "Pandemic Perspectives: The Lost Trials Of 2020" - Scrip, 9 Mar, 2021.)
Davis, who comes with over a decade of experience in the clinical research sector at both leading Indian and foreign firms, also shared his view on India’s Phase III trial waiver clause and underscored the importance of multi-regional clinical trials (MRCTs)as the principal source of evidence across different geographic regions through a framework for understanding ethnic differences in the efficacy and safety of a product.
Significantly, Harvard University’s MRCT Center has been actively engaging in India since 2013 as part of efforts to determine “tractable” solutions to regulatory reform issues. (Also see "Adverse Event Assessment In India Gets Harvard-Built Tool; Will Increased Trial Activity Follow?" - Pink Sheet, 24 Feb, 2017.)
The center, in collaboration with international law firm Ropes & Gray LLP, in 2019 hosted a special presentation and discussion with the Drugs Controller General of India to discuss India’s New Drugs and Clinical Trials Rules, 2019, that were released in March that year.
ISCR also hopes to work cohesively with other stakeholders to develop the ecosystem in India for newer models such as virtual and hybrid trials.
Q: It was a tough 2020, yet the numbers suggest that the Indian trials sector managed to retain some buoyancy - until the middle of November 2020, the Central Drugs Standard Control Organization (CDSCO) approved close to 160 trials, including the 70 global clinical trial (GCTs) applications, compared to 184 trials cleared in all of 2019, of which 96 were GCTs. Was that possible in part on account of the regulatory accommodations provided by CDSCO? (Also see "COVID-19, Lockdown Affect Trials in India But Coping Efforts In Hand" - Scrip, 31 Mar, 2020.) What are the current trends in terms of global sponsor interest in placing studies in India?
A: With the release of the New Drugs and Clinical Trial Rules in 2019, there is good clarity around several issues that were grey areas for global sponsors, like timelines for GCT approvals (90 days), post-trial access, ethics committee registration etc. Global sponsors usually plan their clinical development programs very early and it is difficult for them to change country footprint decisions rapidly before they are certain that the clinical research ecosystem is stable. Increasingly, global sponsors were able to include India as part of the clinical trial footprint from 2020 and this is reflected in the general buoyancy seen in the number of trials being placed.
While the CDSCO embraced digital adoption in a big way in the midst of the pandemic in 2020, like calendarization of Subject Expert Committee meetings which were virtual, faster responses to online submissions in SUGAM [the online portal], etc, at no point has any process for review and approval of submissions deviated from that stated in the rules. India is a significant market globally and global sponsors would definitely want to include the country as part of their global programs as this ensures that Indian patients gets the benefit of participation in research of a new molecule for unmet medical need and, at the same time, it also paves the way for marketing authorization for medicines at the earliest if the medicine/vaccine/medical device has the appropriate safety and efficacy.
Q: How did industry keep trial participation going in India during the tough pandemic year 2020 and also handle the challenges around informed consent of trial participants, given that Indian rules require audio-visual recording of this process?
A: It was a difficult time for all stakeholders of clinical research in 2020. Everyone tried to initially manage the situation as best as possible by invoking business continuity plans where appropriate. Sponsors were quickly able to incorporate and adopt several changes in the conventional way of working, e.g. implementation of remote monitoring, direct-to-participant shipment of medicinal products from the clinical trial site, etc. In the case of sites where trials were ongoing, there was an initial pause during the country-wide lock-down due to trial sites not accessible to patients or due to some sites being converted into COVID-19 care centers.
Investigators worked with ethics committees to best manage ongoing patients and drug supplies, report serious adverse events, manage study visits and so on. In the case of studies with an NCE/NBE, new patients underwent audio video consenting of the informed consent process, especially in non-COVID-19 studies. In the case of COVID-19 studies, investigators worked with ethics committees to seek the best possible way for consenting either by reading out the form and/or audio-videotaping the same with the patient or their ‘Legally Acceptable Representative’ or using an impartial witness. Sponsors also communicated the implementation process to the regulators.
Q: Experts at the recent ISCR conference spoke about the need for increased virtual participation and also hybrid trials with a home nursing component, and greater use of patient engagement apps, given that an estimated 15-30% of participants tend to drop out of trials. Do you expect these models to become mainstay in most markets globally in the post-COVID-19 world and are countries like India moving in the same direction?
A: Yes, models like virtual trials/decentralized trials have been talked about and adopted by several organizations over the past few years globally but it is with the pandemic that this has gained momentum and become more mainstream. While some of the components of virtual/hybrid/decentralized trials are already available in India, there is still a long way to go before these become mainstream and the go-to model for operationalizing clinical trials. There are organizations in India which are already in the home health care, digital health space and this is expected to increase rapidly in breadth and scope in the coming years. ISCR will definitely collaborate with all stakeholders to build the ecosystem for this in India and a lot of it would be in the form of sensitization and socialization of the concepts of virtual/hybrid trials with different stakeholders. A major initiative would be to dispel any misconceptions around these studies, so that everyone involved in clinical trials is comfortable that there is appropriate oversight for these studies and that the same standards of ethics, data quality and patient protection apply.
Q: Artificial intelligence and machine learning have huge scope in optimizing clinical trials across various aspects including study design, effective trial site identification and pharmacovigilance, despite some challenges around electronic medical records (EMR) data mining and data privacy. How far away are we in India from the global median in terms of deploying AI/ML tools in clinical research and in which areas do you see early potential for these smart technologies in developing nations like India?
A: While there have been lot of strides in usage of AL/ML in different domains of clinical research, in India we are also still ramping up (albeit at a slower pace) on EMR systems in hospitals. An additional area of impact is data privacy-related aspects which is still an evolving area and will see a lot of shifts in the coming years as more and more hospitals (government and private sector) adopt EMR as mainstream. In the current scenario, areas like pharmacovigilance, medical writing, drug supply logistics and study/site feasibilities are areas where AL/ML is being used in a tangible manner.
Q: Some experts suggest that India’s Phase III trial waiver clause is a double-edged sword. Global sponsors often pursue that option and at times also seek relaxations in Phase IV requirements subsequently, while some experts claim that trial waiver conditions - such as “no probability or evidence, on the basis of existing knowledge, of any difference in the metabolism of the new drug by the Indian population, or any factor that may affect the pharmacokinetics, pharmacodynamics, and safety and efficacy of the new drug” - are open-ended. Does ISCR see significant value in the trial waiver provisions?
A: Rules 75 and 80 clause (7) of Chapter X of the New Drugs and Clinical Trials Rules, 2019 define the criteria under which an importer/manufacturer can seek a waiver from conducting a local clinical trial prior to grant of a new drug approval in India. ISCR believes that the adoption of these regulations will allow early access to new treatment options to patients in India without compromising patient safety, which is one of the key objectives of the new regulations. Currently there is inconsistent implementation of the rules regarding clinical trial waivers for Phase III or IV. Sponsors are trying to include India in global Phase III studies by planning early. Given that the sample sizes for global studies are shrinking year on year (e.g. an oncology medicine may have a total of 500-600 subjects across Phase III pivotal studies), it is unlikely that India will get significant number of patients on these global programs when there are 30-40 countries who are also vying for inclusion.
With the increasing globalization of drug development, it has become important to consider multi-regional clinical trials [stipulated in guideline E17 of the International Conference on Harmonisation, which describes general principles for the planning and design of MRCTs] as the primary source of evidence across different geographic regions through a framework for evaluating the impact of ethnic factors upon the efficacy or safety of a product. MRCTs can facilitate simultaneous global development of a drug and reduce the number of clinical studies conducted separately in each region, thereby minimizing unnecessary duplication of studies. MRCTs have been recognized as an efficient way to enable recruitment of the planned number of trial subjects within a reasonable time frame when either the disease and/or condition is rare (e.g., an enzyme deficiency disorder), for special (e.g., elderly, pediatric) populations, or when very large numbers of subjects are required from the perspective of proving efficacy and safety (e.g., cardiovascular outcome studies for diabetes drugs, vaccine efficacy studies). It allows for an objective examination of the applicability of a treatment to diverse populations.
The intrinsic and/or extrinsic factors that are believed or suspected to impact upon responses to the drug can be further evaluated based on data from various regions using a single protocol. For example, the impact on the treatment effect of genetic differences or different distribution of gene polymorphisms in drug metabolizing enzymes or the molecular target of a drug can be examined in exploratory and/or confirmatory MRCTs that include subjects with different intrinsic factors across regions.
Accumulated knowledge of the impact of intrinsic and extrinsic factors, and global sharing of experiences in various regions, can facilitate reliance of data generated across the regions. It is also pertinent to note that when data is analysed from global clinical development programs involving multi-regional clinical trials, ethnic variability has not been observed across both several ethnic groups as well as medicines, and it is also postulated that the likelihood of the Indian population alone behaving differently from all other ethnic groups is low. ISCR would also like to emphasize that while member companies may have data from local studies that have been conducted with their own compounds and the data would fall into the above reasoning, it could be possible that the regulatory agency has observed ethnic variability for compounds. The scientific community will be enlightened if this data is published in the public domain.
Q: There have been questions around compliance and data at some CROs in India over the years. Is that broadly a thing of the past especially with the progress made with COVID-19 vaccines and digitization tools and other checks in place helping in this area or does it still weigh on sponsors’ minds while placing studies in India?
A: While ISCR would not be able to comment on individual companies or groups, even if there have been challenges in the past, they were the exception rather than the rule and there are now appropriate control steps and oversight being implemented. ISCR strongly believes that the state of business in India should not be too dissimilar to other countries where clinical trials are conducted. We would like to encourage everyone to focus on the current situation and move away from negative narratives related to events of the past. In light of the more positive changes in the regulatory environment, including the New Drugs and Clinical Trials Rules, 2019, and the need to provide access to new medicines which would not be possible without conducting global clinical trials, both global and local sponsors have put in place robust mechanisms for ensuring that Good Clinical Research Practice is implemented in letter and spirit.
Varying Stages Of Adoption
BY VIBHA RAVI
Agreeing that decentralized trials are here to stay, experts discussed in a recent meeting ways in which the pharma research landscape is set to change. With nearly 85% of the industry at different stages of adoption, such trials have seen greater traction in the US, while stricter data privacy laws have hampered progress in Europe, ISCR’s annual conference heard.
According to some studies, 85% of clinical trials fail to retain enough patients, 70% of participants live over two hours from a research site, and in 50% of studies, participants find it difficult to stay enrolled due to poor health.
No wonder then that one of the most significant changes in the clinical trials landscape during COVID-19 has been greater adoption of decentralized approaches, with even regulators adapting to permit such trials, a recent virtual conference heard.
Q: Finally, looking ahead, how do you see COVID-19 re-shaping the broader clinical research landscape – do you expect the dominance of oncology as a segment for trials to tail off or sharp shifts in the distribution of clinical trial activity globally or a greater digital connect with patients?
A: The pandemic has certainly put more focus on infectious diseases, vaccine and diagnostics as areas which need more research to be carried out. India has a fair share of communicable and non-communicable diseases and would have to balance investments in R&D to ensure that patients are benefited across the spectrum. While investments in oncology R&D will continue to be key for several sponsors, there is also the resurgence of interest in other areas. Patient centricity is a key factor of the future of clinical trials and stakeholders are trying to incorporate the patient voice in clinical trials, not as a tokenism but with a seat at the table for discussions on design of the studies, endpoints, electronic patient-reported outcomes.
One year on from the World Health Organization declaring COVID-19 a global pandemic on 11 March 2020, editors across Informa Pharma Intelligence publications are taking a closer look at its impact and possible lasting implications for the biopharma and medtech industries.
With decentralized trials (DCTs) here to stay, new roles in clinical research are likely to be created, or existing roles expanded, to adapt to the shift from site-focused to patient-centered trials, said panelists at the 14th annual conference of the Indian Society for Clinical Research (ISCR).
“It will be a continuum. Monitors will have access to a lot more data in real time and can play a more proactive approach rather than reactive," said Rajesh Jain, principal consultant clinical solutions at PharmaLex (India), a services provider to pharmaceutical, biotech and medtech industries. "New roles will come up - perhaps a patient support help desk - and there will be a shift from monitoring to patient relationship and support as technology components become more mainstream.”
As use of technology and devices like wearables becomes commonplace, automated data analysis will play an increasingly important role in trials, he predicted.
Levels of adoption of DCTs vary across the world, as regulators are at different stages of preparedness for the change, panelists felt. While the US has been at the forefront, with the Food and Drug Administration having gradually created a regulatory pathway since 2007, the European Medicines Agency has also been proactive. However, data privacy concerns have held back greater adoption in Europe.
Nimita Limaye, research vice-president at International Data Corporation (IDC), said while 52 DCTs are listed on clinicaltrials.org (17 observational and 35 interventional), “it’s a matter of terminology” and the number of such trials is significantly higher in reality.
“I haven’t seen any specific statistics published on this but I feel a significant number of trials that are being approved have a lot of components of decentralization in them,” she added.
Jain said such trials are taking place in Asia-Pacific, including in India, though not all are meant for registration studies or marketing authorization.
For companies looking for a framework to enable DCTs, the panelists advised setting up an internal team with multiple stakeholders to drive the initiative, identify the right technology and consider the therapeutic areas suited to such trials. However, patient centricity is paramount.
“From get-go, it should be an adaptable and scalable approach that tries to keep patients at home," said Isaac Rodriguez-Chavez, senior vice-president, scientific and clinical affairs at PRA Health Sciences, a global contract research organization.
"Not one size fits all – look at the nature of the disease, the target population, which technology needs to be customized, what endpoints need to be measured for the trial to be synchronized with an electronic platform, can the investigational material be delivered safely, are we in compliance with regulations and can you deploy it across different geographic areas, so take a holistic approach,”
While the health journey of a clinical trial participant and technology customization are essential to keep in mind while designing an ecosystem for DCTs, quality assurance, communications plan and data flow also need to be paid heed to, Rodriguez-Chavez observed.
“This reminds me of the title of the book that Dr. Eric Topol wrote, 'The Patient Will See You Now,' and this is what we are really moving towards with patient experience or 'Px.' The technology for the implementation plan has to be truly centered around the patient, keeping patient optionality in mind…so keeping a modular approach. You may have situations where some patients may not be comfortable doing things remotely, while others may love it,” observed IDC’s Limaye.
While adverse or serious adverse events might seem more difficult to monitor in such studies given the virtual nature of interaction between a trial participant and an investigator, it is possible to remotely handle such events, the ISCR meeting heard.
“It’s about the way we set up an alert mechanism and the way we set up a response system for an investigator or a primary care physician to act. We have healthcare delivery systems around - patients are generally in good reach of a system – so how do we build that last mile of connect. We don’t need to build such systems. They exist and it’s only a matter of connecting the right dots,” noted Rajneesh Patil, vice president, Global Site Management at IQVIA in the US.
PharmaLex (India)’s Jain also spoke about the role of "geo-fencing", which is the use of global positioning systems or radio frequency identification technology to create a virtual geographic boundary, thus enabling software to trigger a response when a mobile device enters or leaves a particular area, in creating such alerts.
“A patient is just probably walking into the nearest healthcare center instead of informing the investigator first and we used geo-fencing to create alerts for the investigator. Our patients and investigators found this to be one of the most useful tools,” he added.
The majority of trial participants want the enhanced convenience of a DCT, said PRA Health Science’s Rodriguez-Chavez. “The patient community is very supportive – it’s less disruptive, it's transformative, more inclusive of diverse populations breaking geographic barriers that traditionally existed – for example, in inclusion of people living in rural or difficult to access areas.”
However, IDC’s Limaye said employees at research sites were skeptical due to an underlying fear that such sites might lose relevance and may no longer be required in DCTs.
“But we know now that they are not going away – it’s their operating model that is changing. There was a survey and two-thirds of the sites said they will be very happy to pivot to this model and do whatever it takes to adapt. They are a big piece of what is happening. Though we’re moving from a site-centric to a patient-centric approach, they are still a key component along with technology,” she observed.
However, clinical sites are concerned about access to data and at times technology is seen “as a burden”, given that investigators have not just to train patients on using a device or an application but also need to ensure compliance. While some also feel it would be difficult to establish a “connect” to patients, it could be easier in real life as the effort and time to travel to a site is cut down, Limaye added.
“However, it should be approached on a case-by-case basis as in some serious diseases or conditions, empathy and a physical connect is required. Significant pieces of DCTs are here to stay, but it won’t apply to everything.”